Cat.NO.:A256160,A2670810,A256160,A2670810 Purity:98%,99%,98%,99%
Product Details of 3-Hydroxybenzaldehyde
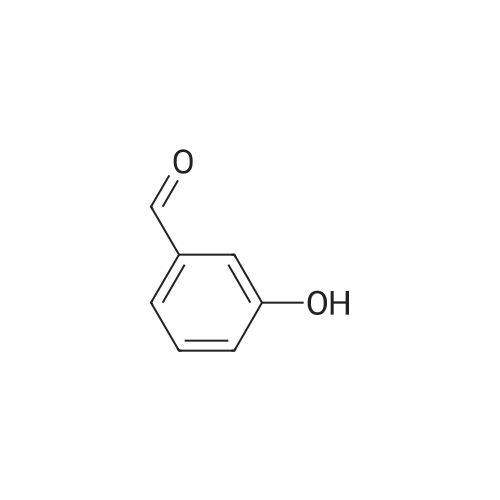
| CAS No. : | 100-83-4 |
| Formula : |
C7H6O2 |
| M.W : |
122.12
|
| SMILES Code : | O=CC1=CC=CC(O)=C1 |
| Synonyms : |
M-Hydroxybenzaldehyde; 3-HBA
|
| MDL No. : | MFCD00003368 |
Safety of 3-Hydroxybenzaldehyde
| GHS Pictogram: |  |
| Signal Word: | Warning |
| Hazard Statements: | H315-H319-H335 |
| Precautionary Statements: | P261-P305+P351+P338 |
Application In Synthesis of 3-Hydroxybenzaldehyde
* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
- Upstream synthesis route of [ 100-83-4 ]
- Downstream synthetic route of [ 100-83-4 ]
[ 100-83-4 ] Synthesis Path-Upstream 1~4
- 1
 [ 100-83-4 ]
[ 100-83-4 ]
 [ 20035-32-9 ]
[ 20035-32-9 ]
| Yield | Reaction Conditions | Operation in experiment |
|---|---|---|
| 35.5 g | With bromine In dichloromethane; water at 0℃; Inert atmosphere | Reaction step: 35.4 g of 3-hydroxybenzaldehyde and 212 ml of dichloromethane were added to a clean reaction vessel, and the mixture was replaced with vacuum nitrogen three times, stirred and cooled to below 0 ° C, 35.4 g of liquid bromine was added dropwise, and 250 ml of water was added after completion of the reaction. Adjust the pH to neutral with liquid alkali, filter by cooling, add 354ml of wet product toluene to dissolve, separate the water layer, warm the organic phase to reflux, filter by temperature, and dry to obtain 4-bromo-3-hydroxy-benzaldehyde 35.5. g. |
[1] Synthetic Communications, 2010, vol. 40, # 5, p. 647 – 653.
[2] Bioorganic and Medicinal Chemistry, 2014, vol. 22, # 4, p. 1285 – 1302.
[3] Patent: CN109053443, 2018, A, . Location in patent: Paragraph 0025; 0034; 0051-0054; 0059; 0074-0077; 0097-0100.
- 2
- [ 100-83-4 ]
 [ 2973-80-0 ]
[ 2973-80-0 ]
 [ 3111-51-1 ]
[ 3111-51-1 ]- [ 20035-32-9 ]
[1] Tetrahedron, 2010, vol. 66, # 34, p. 6928 – 6935.
- 3
- [ 100-83-4 ]
- [ 2973-80-0 ]
- [ 20035-32-9 ]
[1] Chemistry – A European Journal, 2007, vol. 13, # 30, p. 8543 – 8563.
- 4
- [ 100-83-4 ]
 [ 1206800-24-9 ]
[ 1206800-24-9 ]
[1] Organic Process Research and Development, 2014, vol. 18, # 4, p. 501 – 510.
[ 100-83-4 ] Synthesis Path-Downstream 1~35
- 1
 [ 75-30-9 ]
[ 75-30-9 ]- [ 100-83-4 ]
 [ 75792-33-5 ]
[ 75792-33-5 ]
| Yield | Reaction Conditions | Operation in experiment |
|---|---|---|
| 95% | With potassium carbonate; In N,N-dimethyl-formamide; at 50℃; for 6h; | 3-hydroxybenzaldehyde (61A) (3.05 g, 25.0 mmol)Dissolved in N,N-dimethylformyl (30mL), and then Potassium carbonate (6.9 g, 50.0 mmol) and iodoisopropane(5.09 g, 30.0 mmol) was added to the reaction, and the reaction was carried out at 50 C for 6 hours. The reaction was cooled to 0 C and water (90 mL) was added.The aqueous phase was extracted with methyl tert-butyl ether (30 mL×3) and the organic phases were combined.Wash with saturated sodium chloride (30 mL × 1),Dry over anhydrous sodium sulfate, filter, and concentrate the filtrate to give3-Isopropoxybenzaldehyde (61B) (3.9 g, yield: 95%). |
| 85% | With potassium carbonate; In N,N-dimethyl-formamide; for 42h; | A solution of 3-hydroxybenzaldehyde (67 g, 549 mmol), 2-iodopropane (100 g, 588 mmol) and K2CO3 (130 g, 942 mmol) in 400 mL DMF was stirred for 18 h. To the above solution was added 2-iodopropane (20 g, 117 mmol) and K2CO3 (20 g, 145 mmol) and the mixture was stirred for another 24 h. The reaction mixture was poured into 300 mL water which was extracted with EtOAc (200 mL×3). The combined organic layers were washed with water (100 mL×3), dried over Na2SO4, filtered, and concentrated in vacuo to afford 77 g (85% yield) of 3-isopropoxybenzaldehyde as an oil. 1H NMR (300 MHz, CDCl3) δ 1.29-1.31 (d, 6H) 4.5-4.6 (m, 1H) 7.0-7.1 (m, 1H) 7.3-7.4 (m, 3H). |
| 71% | With potassium carbonate; In ethanol; at 60℃; for 22h;Inert atmosphere; | This method of alkylation is similar to that described in the Journal of Medicinal Chemistry (2002), 45(18), 3891-3904. Dissolved 3-hydroxy benzaldehyde (0.500 g, 4.094 mmole) in 10 mL EtOH at room temperature under argon, and magnetically stirred. Added K2CO3. (1.132 g,8.189 mmole), then 2-iodopropane (0.819 mL, 8.819 mmole). Warmed to 60 C. After 18 hours, added a second equivalent of alkyl iodide (0.819 mL, 8.189 mmole) and continued heating at 60 C. After 4 hours, cooled to room temperature. Concentrated in vacuo. Partitioned residue between EtOAc, and H2O. Washed organic layer with 1 M NaOH, 2x 20 mL, dried organic layer over MgSO4, filtered and concentrated in vacuo. Yield 0.48g, 71%.The material was taken on immediately to the next step. |
| 62% | 172 (1.99 g, 16.3 mmol) was added to a solution of K2CO3(4.08 g, 29.5 mmol) in acetone (40 mL) at 0 C under Ar, then thereaction was stirred for 15 min. After 2-iodopropane (3.32 mL,32.8 mmol) was added to the solution, the resulting mixture washeated until reflux began this being maintained for 9 h. The reactionwas quenched with H2O (20 mL) and extracted with EtOAc(3 x 30 mL). The organic layers were washed with H2O (20 mL)and brine (20 mL), dried (MgSO4), filtered and concentrated in vacuo.The residue was purified by silica gel CC (EtOAc/hexane,10:90) to afford 3-isopropoxybenzaldehyde (76) (1.61 g,9.78 mmol, 62%) as a yellow oil: 1H-NMR (CDCl3, 400 MHz) d:1.36 (d, J = 5.6 Hz, 6H, -CH3), 4.63 (heptet, J = 5.6 Hz, 1H, -CH-(CH3)2), 7.15 (m, 1H, Ar-H), 7.38 (brs, 1H, Ar-H), 7.42-7.43 (m,2H, Ar-H), 9.97 (s, 1H, -CHO); spectral data were in agreementwith those in the literature (Garca-Daz et al., 2011). | |
| N-(2-(3-Isopropoxy)phenethyl)-N’-(2-thiazolyl)thiourea 3-Isopropoxybenzaldehyde was prepared from 3-hydroxybenzaldehyde and isopropyl iodide analogous to the procedure described in Example 361. | ||
| With potassium carbonate; In ethanol; at 60℃; for 22h; | This method of alkylation is similar to that described in J Med Chem (2002), 45(18), 3891-3904. Dissolved 3-Hydroxy benzaldehyde (0.500g, 4.094 mmole) in 10 mL EtOH at rt, under Argon, and magnetically stirred. Added K2CO3. (1.132g, 8.189mmole), then 2-iodopropane (0.819 mL, 8.819 mmole). Warmed to 6O0C. After 18 hr, added a second equivalent of alkyl iodide of (0.819 mL, 8.189 mmole) and continued heating at 6O0C. After 4 hr, cooled to rt and concentrated in vacuo. Partitioned residue between EtOAc, and H2O. Washed organic layer with 1 M NaOH, 2x 2OmL , dried organic layer over MgSO4, filtered and concentrated in vacuo to give the aldehyde. | |
| With potassium carbonate; In isopropyl alcohol; for 8h;Heating / reflux; | Step 10a: 3-isopropoxybenzaldehyde; To a 1 molar solution of 3-hydroxybenzaldehyde (10Og, 0.819 moles) in isopropyl alcohol (820 mL) was added 2-iodopropane (146.2g, 0.860 moles) followed by powdered potassium carbonate (339.5g, 0.457 moles) and the mixture was heated to reflux under nitrogen for a minimum of 8 hrs. The reaction was complete by TLC. Water was added to the cooled reaction until all salts were dissolved. The mixture was extracted with ether three times. The combined ether extracts were washed with water, 2 M NaOH and again with water until clear (three times) then brine. The organic layer was dried over magnesium sulfate and filtered and evaporated to give the desired product as a pale oil. lH NMR (400 MHz, CDC13) δ 9.95 (s, IH), 7.2 (m, 3H), 7.12 (m, IH), 4.6 (m, IH), 1.32 (bs, 6H) ppm. LCMS ion fragment = 164.1 | |
| With potassium carbonate; In isopropyl alcohol; for 8h;Heating / reflux; | 3-lsopropoxy-benzaldehvde.A solution of 3-hydroxybenzaldehyde (4.5 g) and 2-iodopropane (3.72 ml_) in 2-propanol (40 mL) was treated with K2CO3 (16.09 g). After 8 h at reflux, water (40 mL) was added and the mixture was extracted with Et2θ (3x 25mL). The combined organic layers were washed with water (25 mL), 2 M NaOH (25 mL), water (4×25 mL), and satd. aq. NaCI (25 mL). The organic phase was dried (Na2SO4), and concentrated to give the title compound as a pale orange oil (3.31 g). 1H NMR (CDCI3): 9.96 (s, 1H), 7.45-7.41 (m, 2H), 7.38-7.37 (m, 1H), 7.17-7.13 (m, 1 H), 4.68-4.59 (septet, 1 H1 J= 6.1 Hz), 1.37-1.35 (d, 6H, J= 6.1 Hz). | |
| With sodium carbonate; In N,N-dimethyl-formamide; at 50℃; for 100h; | A 75 mL mini-block tube equipped with a magnetic stirrer bar was charged with 6.16 g (50.4 mmol) of 3-hydroxybenzaldehyde, 4.40 mL (8.58 g, 55.0 mmol) of isopropyl iodide 30 mL of dimethylformamide and 6.05 g (57.1 mmol) of sodium carbonate. The slurry was heated to 500C.After 40 hours the reaction was only 50% complete so another 3 ml (5.85 g, 37.4 mmol) isopropyl iodide was added. After 60 more hours another 3 mL (5.85 g, 37.4 mmol) isopropyl iodide and 3 g (28.5 mmol) of sodium carbonate were added. The reaction mixture was cooled to 25C and diluted with methyl t-butyl ether (MTBE) and water. The organic layer was decanted off. The aqueous phase was rinsed with MTBE, again decanting off the organic layer. The combined organic layers were washed with 2N aqueous sodium hydroxide (3 X 30 mL) and brine (1 X 30 mL), dried over sodium sulfate and concentrated to give 3-Isopropoxybenzaldehyde which was used as (following the above procedure for 3-(4-t-butylphenyl)propanoic acid (Example 6) to prepare 3-(3- Isopropoxyphenyl)propanoic acid (1.31 g) as an off-white solid. IH NMR (d6-DMSO): δ 12.0, bs, IH (COOH); δ 7.03, t, IH, (arylH meta to O-i-Pr); δ 6.6, m, 3H (other arylH’s); δ 4.45, hept, IH, (OCH); 2.65, t, 2H, (CH2 α to aryl); δ 2.38, t, 2H (CH2 α to COOH); δ 1.12, d, 6H (CH3’s). 13C NMR (d6-DMSO): 173.70, 157.40, 142.46, 129.26, 120.17, 115.54, 112.93, 68.77, 35.09, 30.31, 21.84. |

[2]Patent: US2008/194535,2008,A1 .Location in patent: Page/Page column 36-37.
[3]Bioorganic and Medicinal Chemistry Letters,2015,vol. 25,p. 4812 – 4819.
[4]Journal of Medicinal Chemistry,2002,vol. 45,p. 3891 – 3904.
[5]Phytochemistry,2013,vol. 96,p. 132 – 147.
[6]Journal of the Chemical Society,1955,p. 461,464.
Journal of the Chemical Society,1959,p. 1027,1030.
[7]Patent: US5593993,1997,A .
[8]Patent: WO2006/44497,2006,A2 .Location in patent: Page/Page column 55.
[9]Patent: WO2007/11810,2007,A1 .Location in patent: Page/Page column 37; 42.
[10]Patent: WO2007/140005,2007,A2 .Location in patent: Page/Page column 28.
[11]Patent: WO2008/112368,2008,A2 .Location in patent: Page/Page column 35.
- 2
- [ 100-83-4 ]
- [ 106-95-6 ]
 [ 40359-32-8 ]
[ 40359-32-8 ]
| Yield | Reaction Conditions | Operation in experiment |
|---|---|---|
| 100% | With potassium carbonate; sodium iodide; In ethanol; for 3h;Reflux; | Example 1 . Preparation of 3-Allyloxybenzaldehyde; [0021] In a 1 L round bottomed flask equipped with mechanical stirrer, reflux condenser and thermometer were added 400 ml_ ethanol, 59.63 g of 3- hydroxybenzaldehyde (0.49 moles, 1 eq.), 7.3 g of sodium iodide (48 mmol, 0.1 eq.), 120.98 g of allyl bromide (0.59 moles, 1 .2eq.) and 101 .6 g of potassium carbonate (0.74 moles, 1 .25 eq.). The reaction mixture was heated to reflux and heating continued for three hours. Heating was then discontinued and the reaction was allowed to cool to room temperature. The mixture was then filtered through a Hyflosupercel pad and ethanol was removed by rotary evaporation. The residual oil was then taken up in 500 ml_ of MTBE and the organic phase washed sequentially with 10% aqueous sodium hydroxide, water and brine. After drying over sodium sulfate, filtration and rotary evaporation of solvent 79.7 g of a yellow oil of 3-allyloxybenzaldehyde (quantitative yield) was obtained. |
| 100% | With potassium carbonate; In ethanol; for 3h;Reflux; | Example 1 . Preparation of 3-Allyloxybenzaldehyde 3; [0016] In a 1 L round bottomed flask equipped with mechanical stirrer, reflux condenser and thermometer was added in sequence 400 mL ethanol, 59.63 g of 3-hydroxybenzaldehyde (0.49 moles, 1 eq.), 7.3 g of sodium iodide (48 mmol, 0.1 eq.), 120.98 g of allyl bromide (0.59 moles, 1 .2eq.) and 101 .6 g of potassium carbonate (0.74 moles, 1 .25 eq.). The reaction mixture was heated to reflux and heating continued for three hours. Heating was then discontinued and the reaction was allowed to cool to room temperature. The mixture was then filtered through a Hyflosupercel pad and ethanol was removed by rotary evaporation. The residual oil was then taken up in 500 mL of MTBE and the organic phase washed sequentially with 10% aqueous sodium hydroxide, water and brine. After drying over sodium sulfate, filtration and rotary evaporation of solvent 79.7 g of a yellow oil of 3-allyloxybenzaldehyde (quantitative yield) was obtained. |
| 100% | With potassium carbonate; sodium iodide; In ethanol; for 3h;Reflux; | In a 1 L round bottomed flask equipped with mechanical stirrer, reflux condenser and thermometer were added 400 mL ethanol, 59.63 g of 3-hydroxybenzaldehyde (0.49 moles,1 eq.), 7.3 g of sodium iodide (48 mmol, 0.1 eq.), 120.98 g of allyl bromide (0.59 moles,1.2 eq.) and 101.6 g of potassium carbonate (0.74 moles,1.25 eq.). The reaction mixture was heated to reflux and heating continued for three hours. Heating was then discontinued and the reaction was allowed to cool to room temperature. The mixture was then filtered through a Hyflosupercel pad and ethanol was removed by rotary evaporation. The residual oil was then taken up in 500 mL of MTBE and the organic phase washed sequentially with 10% aqueous sodium hydroxide, water and brine. After drying over sodium sulfate, filtration and rotary evaporation of solvent 79.7 g of a yellow oil of 3-allyloxybenzaldehyde (quantitative yield) was obtained. |
| 98% | With potassium carbonate; In N,N-dimethyl-formamide; at 20℃;Inert atmosphere; | General procedure: To a suspension ofphenol (16, 17, 22, and 23) (1 mmol, 1.0 equiv) and K2CO3(3 mmol, 3 equiv) in anhydrous DMF(5 mL) was added allylbromide (1.5 mmol, 1.5 equiv) dropwise under nitrogenatmosphere at room temperature. The mixture was stirred atroom temperature for 1 h. After completion of the reaction,water (10 mL) was added. The mixture was then extractedwith ether (2 × 30 mL), the combined organic layer waswashed with brine (2 × 30 mL), dried over anhydrousNa2SO4, filtered and the filtrate was concentrated in vacuo.The crude residue was purified by column chromatography (EtOAc/hexane = 1/8) to yield the allyl protected aldehyde(11, 18, 7, and 9). |
| 98% | With potassium carbonate; In N,N-dimethyl-formamide; at 20℃; for 1h;Inert atmosphere; | Phenol (16, 17, 22 and 23) (1 mmol, 1.0 eq.) And Allyl bromide (1.5 mmol, 1.5 eq.) Was added dropwise to a suspension of K2CO3 (3 mmol, 3 eq.) In anhydrous DMF (5 mL) under nitrogen atmosphere at room temperature. The mixture was stirred at room temperature for one hour. After completion of the reaction, water (10 ml) was added. The mixture was extracted with ether (2 x 30 mL) and the organic layer was washed with brine (2 x 30 mL), dried over anhydrous Na2SO4, filtered and the filtrate was concentrated in vacuo. The crude compound was purified by column chromatography (EtOAc / hexane = 1/8) to give aldehydes 11, 18, 7 and 9 protected with allyl groups. |
| 98% | With 18-crown-6 ether; potassium carbonate; potassium iodide; In acetonitrile; at 75℃; for 18h; | To a stirred solution of 3-hydroxybenzaldehyde (8.0 g, 65.6 mmol) in acetonitrile (80 mL) was added allylbromide (11.0 mL, 131 mmol), KI (1.09 g, 6.56 mmol), 18-crown-6 (864 mg, 3.26 mmol) and K2CO3 (26.4 g, 191 mmol) at rt, after which the solution was refluxed at 75 C for 18 h. The K2CO3 was filtered off and the solvent was removed under reduced pressure·H2O (100 mL) was added and the aqueous layer extracted with EtOAc (2 * 100 mL). The organic layer was separated, washed with brine (50 mL), dried (Na2SO4) and the solvent removed under reduced pressure to yield the title compound as an orange oil, which was used without further purification (10.5 g, 64.4 mmol, 98%). Rf = 0.38 (5% EtOAc in hexane). IR: lambdamax = 2861 (w, C-H), 1681 (s, C=O), 1598 (s, C=C), 1483 (m, C=C), 1457 (m, C=C). 1H NMR (400 MHz, CDCl3): deltaH = 9.92 (1H, s, H10), 7.37-7.44 (2H, m, H6 & H7), 7.36 (1H, dd, J = 2.0, 1.0 Hz, H9), 7.15 (1H, dt, J = 7.2, 2.4 Hz, H5), 5.96-6.09 (1H, ddt, J = 17.2, 10.5, 5.2 Hz, H2), 5.40 (1H, dq, J = 17.3, 1.5 Hz, H1t), 5.28 (1H, dq, J = 10.6, 1.4 Hz, H1c), 4.55 (2H, dt, J = 5.3, 1.4 Hz, H3). 13C NMR (101 MHz, CDCl3): deltaC = 192.0 (C10), 159.1 (C4), 137.8 (C8), 132.7 (C2), 130.1 (C6), 123.5 (C7), 122.0 (C5), 118.0 (C1), 113.1 (C9), 68.9 (C3) . HRMS (ESI+): m/z found [M+H]+ 163.0751, C10H11O2 required 163.0754. |
| 94.8% | With potassium carbonate; In N,N-dimethyl-formamide; at 25 – 45℃; | Meta hydroxy benzaldehyde (500 g) and potassium carbonate (679 g) were added to DMF (1000 ml). Allyl bromide (569.0 g) was gradually added to the reaction mixture, which was stirred at 25-45C. (0113) After completion of the reaction, water was added to the reaction mass followed by extraction with methyl tertiary butyl ether (MTBE). The organic layer was separated, washed with 1% sodium hydroxide solution (49 g NaOH in 490 ml water) to give compound (2). Yield: 630 g (94.8 %) |
| 92% | With sodium perborate; In water; at 20℃; for 8.5h;Green chemistry; | General procedure: To the 25 mL round bottomed flask containing 5 mL of water, were added beta-Naphthol (1d, 0.50 g, 1 eq.), sodium perborate (3, 0.214 g, 0.4 eq.), and allyl bromide (2, 0.462 g, 1.1 eq.) and reaction mixture was stirred at room temperature as given time. Progress of the reaction was monitored by TLC. After completion of the reaction (6h), the product was extracted with ethyl acetate (3×15 mL) and combined organic layers were dried over anhydrous sodium sulfate and concentrated under reduced pressure. The crude residue thus obtained was purified by silica gel column chromatography (100-200 mesh) to afford pure compound 4d. Similar procedure was adopted for the compounds 4a-c, and 4e-j. |
| 89% | With caesium carbonate; In N,N-dimethyl acetamide; ethyl acetate; | EXAMPLE 58A 3-(allyloxy)benzaldehyde 3-Hydroxybenzaldehyde (1.00 g, 8.19 mmol) and allyl bromide (0.780 mL, 9.01 mmol) in N,N-dimethylacetamide (35 mL) was treated with Cs2CO3 (4.01 g, 12.3 mmol) and allowed to stir at room temperature for 12 hours. The reaction mixture was diluted with diethyl ether (150 mL), washed with 1N HCl, saturated NaHCO3, brine, dried (MgSO4), filtered and the filtrate concentrated. The residue was purified by flash chromatography (silica gel, using 7% ethyl acetate/hexanes as eluent) to provide the title compound as a clear oil (1.19 g, 89%). 1H NMR (CDCl3, 300 MHz) delta 4.82 (m, 2H), 5.25-5.5.50 (m, 2H), 6.03 (m, 1H), 7.18-7.52 (m, 4H), 9.95 (s, 1H); MS (DCI+) 163 (M+H)+. |
| 83% | With potassium carbonate; In N,N-dimethyl-formamide; at 20℃; for 3h; | General procedure: To a solution of 3-hydroxybenzaldehyde 1a (81.9 mmol, 10.0 g) and K2CO3 (1.5 g) in DMF (100.0 mL) was added allyl bromide (7.8 mL) and the mixture was stirred for 3 h at rt. The reaction mixture was quenched with water (50 mL) and the organic phase was extracted into Et2O (3×100 mL), dried over Na2SO4 and the solvent removed at reduced pressure to give a yellow liquid 2a (11.0 g, 83%): FTIR (KBr) 3071, 2818, 1697 cm-1; 1H NMR (200 MHz): delta 9.91 (s, 1H), 7.41 (m, 3H), 7.17 (m, 1H), 6.04 (m, 1H), 5.41 (dd, J 16.0, 6.0 Hz, 1H), 5.30 (dd, J 10.0, 6.0 Hz, 1H), 4.58 (d, J 5.6 Hz, 2H); 13C NMR (50 MHz, CDCl3): delta 191.7, 159.1, 137.7, 132.8, 130.1, 123.6, 122.1, 118.0, 113.1, 68.9; MS(EI) m/z: 162 (M+, 100), 147 (47), 121 (94), 65 (81). |
[2]Patent: WO2012/9816,2012,A1 .Location in patent: Page/Page column 9-10.
[3]Patent: WO2012/9818,2012,A1 .Location in patent: Page/Page column 6.
[4]Patent: US2013/331593,2013,A1 .Location in patent: Paragraph 0032; 0033.
[5]Bulletin of the Korean Chemical Society,2015,vol. 36,p. 2907 – 2914.
[6]Patent: KR2016/115127,2016,A .Location in patent: Paragraph 0044; 0045.
[7]Bioorganic and Medicinal Chemistry,2017,vol. 25,p. 2825 – 2843.
[8]Organic Letters,2018,vol. 20,p. 7011 – 7014.
[9]Patent: WO2017/130109,2017,A1 .Location in patent: Page/Page column 25.
[10]European Journal of Organic Chemistry,2018,vol. 2018,p. 5605 – 5614.
[11]Organic and Biomolecular Chemistry,2019,vol. 17,p. 9489 – 9501.
[12]Journal of the American Chemical Society,1985,vol. 107,p. 1421 – 1423.
[13]Synthetic Communications,2015,vol. 45,p. 355 – 362.
[14]Patent: US2002/173665,2002,A1 .
[15]Chemical Communications,2012,vol. 48,p. 3303 – 3305.
[16]Journal of Chemical Research, Miniprint,1981,p. 2564 – 2572.
[17]Arkivoc,2017,vol. 2017,p. 194 – 209.
[18]Tetrahedron,2000,vol. 56,p. 1873 – 1882.
[19]Patent: US2667509,1951, .
[20]Il Farmaco,2003,vol. 57,p. 909 – 916.
[21]Journal of Organic Chemistry,2003,vol. 68,p. 1146 – 1149.
[22

Reviews
There are no reviews yet.