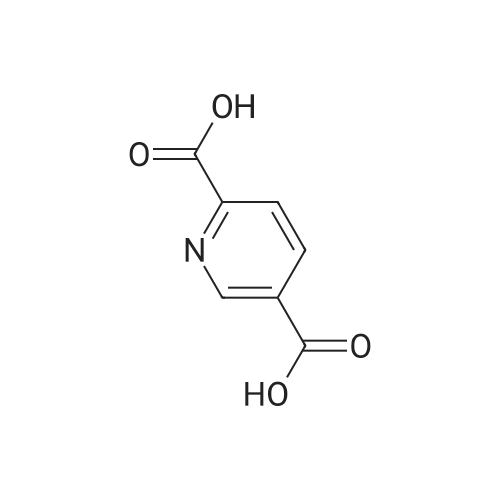
Cat.NO.:A177818 Purity:98%
Product Details of 2,5-Pyridinedicarboxylic acid
| CAS No. : | 100-26-5 |
| Formula : |
C7H5NO4 |
| M.W : |
167.12
|
| SMILES Code : | OC(=O)C1=CN=C(C=C1)C(O)=O |
| MDL No. : | MFCD00006297 |
| InChI Key : | LVPMIMZXDYBCDF-UHFFFAOYSA-N |
| Pubchem ID : | 7493 |
Safety of 2,5-Pyridinedicarboxylic acid
| GHS Pictogram: |  |
| Signal Word: | Warning |
| Hazard Statements: | H315-H319-H335 |
| Precautionary Statements: | P261-P305+P351+P338 |
Application In Synthesis of 2,5-Pyridinedicarboxylic acid
* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
- Upstream synthesis route of [ 100-26-5 ]
- Downstream synthetic route of [ 100-26-5 ]
[ 100-26-5 ] Synthesis Path-Upstream 1~2
- 1
 [ 104-90-5 ]
[ 104-90-5 ]
 [ 3222-47-7 ]
[ 3222-47-7 ]
 [ 770-08-1 ]
[ 770-08-1 ] [ 100-26-5 ]
[ 100-26-5 ]
[1] Pharmaceutical Chemistry Journal, 1993, vol. 26, # 11-12, p. 894 – 896.
[2] Pharmaceutical Chemistry Journal, 1993, vol. 26, # 11-12, p. 894 – 896.
- 2
- [ 100-26-5 ]
 [ 323578-38-7 ]
[ 323578-38-7 ]
[1] European Journal of Inorganic Chemistry, 2010, # 13, p. 1913 – 1928.
[ 100-26-5 ] Synthesis Path-Downstream 1~13
- 1
- [ 100-26-5 ]
 [ 64-17-5 ]
[ 64-17-5 ] [ 5552-44-3 ]
[ 5552-44-3 ]
| Yield | Reaction Conditions | Operation in experiment |
|---|---|---|
| 75% | With sulfuric acid; for 18.0h;Reflux; | Concentrated sulfuric acid (3.9 mL, 71.6 mmol) was added dropwise over 15 minutes to a stirred suspension of 2,5-pyridinedicarboxylic acid (3.0 g, 17.9 mol) in absolute ethanol (10 mL) and the resulting mixture heated to reflux for 18 hours. The solution was allowed to cool to room temperature and the solvents evaporated under reduced pressure. Saturated NaHCC>3 solution was added to the residue to adjust the pH to -8 then the aqueous phase was extracted with EtOAc (4 x 50 mL). The combined organic extracts were washed with brine (30 mL), dried (Na2S04) and evaporated under reduced pressure to leave the title compound (3.0 g, 75%) which was used without further purification. |
| 67% | With sulfuric acid; for 4.0h;Reflux; | Diethyl pyridine-2,5-dicarboxylate (5.3)Pyridine-2,5-dicarboxylic acid (20.0 g, 120 mmol) was dissolved in ethanol (200 mL) and treated with concentrated solution of sulfuric acid (2.0 mL, 36 mmol) in ethanol (20 mL). The reaction was heated under reflux for 4hrs, concentrated and dissolved in EtOAc (10 mL). The mixture was washed with water (50mL), brine (50 mL), dried over sodium sulfate, filtered and evapourated to afford diethyl pyridine-2,5-dicarboxylate (18 g, 67 %). 1 H NMR (400MHz CDCI3) δΗ ppm 1 .25 (t, 3H), 4.3 (q, 2H), 8.2 (d, 1 H), 8.4 (d, 1 H), 9.4 (s, 1 H). |
| 49% | With sulfuric acid; for 48.5h;Reflux; | To a suspension of 2, 5-pyridinedicarboxylic acid (20 g, 120 mmol) in absolute EtOH (120 ml_) was added cone. H2SO4 (25.6 ml_, 0.048 mmol) dropwise over a period of 30 min. The resulting reaction mixture was refluxed for 48 h. The reaction mixture was concentrated, and the resulting residue basified to pH 8 (sat. aq. NaHCCh). The resulting aqueous layer was extracted with EtOAC (4 x 200 ml_). The combined organic layers were washed with brine, washed, dried (Na2SC>4) and concentrated. Four other 20 g batches were reacted in parallel and the resulting crude material from each reaction was combined and purified by column chromatography on silica gel (60-120 mesh) eluting with 5% EtOAC/hexane to afford the title compound (65 g, 291 mmol, 49%). |
| With thionyl chloride; triethylamine; | a) Add 50 mL of thionyl chloride to 25 g of pyridene-2,5-dicarboxylic acid and heat the so-formed mixture at reflux for 6 hrs. Remove all volatiles by vacuum distillation to obtain crude di acid chloride. Immerse flask contain diacid chloride in an ice-batch and add 100 mL of absolute ethanol thereto. Slowly add 30 mL of triethlamine to the stirred reaction mixture and continue stirring for 1/2 hr. Add Ethyl acetate/water to stirred reaction mixture and separate the organic layer. Remove the solvent to produce crude product. Re-crystallize crude product from hexane/methylene chloride to give 25 g of the pyridine-2,5-bis(ethylcarboxylate). | |
| 26.61 g | With sulfuric acid; In water; at 78℃; for 58.0h; | (0201) 2,5-pyridinedicarboxylic acid (25.08 g; 150 mmol) was added to ethanol (1,800 mL). Aqueous sulfuric acid (1.32 g) was added. The mixture was heated at reflux (about 78 C.) for 58 hours, during which time water was removed from the reaction. The reaction progress was monitored using NMR spectroscopy. After the 2,5-diethyl-2,5-pyridinedicarboxylate had been formed in >97% purity by NMR, the reaction mixture was allowed to cool to ambient temperature and was extracted with 2-methyltetrahydrofuran. The combined organic layers were washed with a saturated aqueous brine solution and deionised water, and dried (MgSO4). The organics were filtered and the volatiles were removed in vacuo to afford the title compound (26.61 g; 120 mmol; >99% conversion by GC). |

[2]Journal of Porphyrins and Phthalocyanines,2010,vol. 14,p. 469 – 480.
[3]Dalton Transactions,2015,vol. 44,p. 15391 – 15395.
[4]European Journal of Organic Chemistry,2010,p. 174 – 182.
[5]CrystEngComm,2011,vol. 13,p. 2915 – 2922.
[6]Patent: WO2018/197714,2018,A1 .Location in patent: Page/Page column 115; 116.
[7]Liebigs Annalen der Chemie,1981,p. 2164 – 2179.
[8]Journal of Medicinal Chemistry,1983,vol. 26,p. 1282 – 1293.
[9]Patent: WO2013/72903,2013,A1 .Location in patent: Page/Page column 67.
[10]Patent: WO2021/58754,2021,A1 .Location in patent: Page/Page column 116-117.
[11]Zhurnal Prikladnoi Khimii,1959,vol. 32,p. 2820; engl. Ausg. S. 2904.
[12]European Journal of Medicinal Chemistry,2004,vol. 39,p. 889 – 895.
[13]Angewandte Chemie – International Edition,2015,vol. 54,p. 14570 – 14574.
Angew. Chem.,2015.
[14]Patent: US5350772,1994,A .
[15]Patent: US2018/148752,2018,A1 .Location in patent: Paragraph 0200-0201.
- 2
- [ 100-26-5 ]
- [ 71-36-3 ]
 [ 6938-06-3 ]
[ 6938-06-3 ]
- 3
- [ 100-26-5 ]
- [ 5552-44-3 ]
- 4
- [ 589-93-5 ]
 [ 4434-13-3 ]
[ 4434-13-3 ]- [ 3222-47-7 ]
- [ 100-26-5 ]
- 5
- [ 100-26-5 ]
 [ 4506-66-5 ]
[ 4506-66-5 ]- poly[(1,7-dihydrobenzo{1,2-d:4,5-d’}-bisimidazole-2,6-diyl)-2,5-pyridine]; monomer: 1,2,4,5-tetraminobenzene tetrahydrochloride; monomer: 2,5-pyridinedicarboxylic acid [ No CAS ]
- 6
- zinc(II) sulfate heptahydrate [ No CAS ]
- [ 100-26-5 ]
 [ 4054-67-5 ]
[ 4054-67-5 ]- [ 1174335-78-4 ]
- 7
- [ 100-26-5 ]
- [ 323578-38-7 ]
- 8
- [ 100-26-5 ]
 [ 10165-86-3 ]
[ 10165-86-3 ]
- 9
- [ 100-26-5 ]
- lanthanum(III) nitrate hexahydrate [ No CAS ]
 [ 865169-07-9 ]
[ 865169-07-9 ]- La(3+)*C20H11N4O2(1-)*C7H3NO4(2-) [ No CAS ]
| Yield | Reaction Conditions | Operation in experiment |
|---|---|---|
| 67% | With sodium hydroxide; In water; at 179.84℃; for 72h;pH 8 – 9;Autoclave; High pressure; | General procedure: PrCl3·7H2O (0.1mmol), HNCP (0.1mmol) and 2,5-H2pydc (0.1mmol) were mixed in 10ml deionized water. And its pH value was controlled in the range of 8-9 with 1mol·L-1 NaOH solution. The resulting precursor was sealed in 25ml Teflon-lined stainless steel reactor and heated at 453K for 3d under autogenous pressure. The reaction system was then cooled slowly to room temperature. The obtained solid is a mixture of yellow block crystals and powder. The crystals of 1 suitable for X-ray single-crystal diffraction analysis are picked out from the solid mixture in 61% yield based on PrCl3·7H2O. Syntheses of [Ln(NCP)(2,5-pydc)]n (Ln=La(2), Nd(3), Sm(4)). The complexes 2, 3 and 4 were prepared as described as for 1 above, except that Ln(NO3)3·6H2O was used instead of PrCl3·7H2O. The crystals of complexes 2-4 are picked out from the solid mixture in 67%, 65% and 72% yield based on Ln(NO3)3·6H2O, respectively. Anal. Calcd for C54H34N10O15Pr2 (1): C 48.23, H 2.55, N 10.42%. Found: C 48.01, H 2.63, N 10.10%. Anal. Calcd for C27H14N5O6La (2): C 50.41, H 2.19, N 10.89%. Found: C 50.13, H 2.33, N 10.84%. Anal. Calcd for C27H14N5O6Nd (3): C 49.99, H 2.18, N 10.80%. Found: C 50.03, H 2.25, N 10.77%. C27H14N5O6Sm (4): C 49.53, H 2.16, N 10.70%. Found: C 49.47, H 2.20, N 10.67%. |
- 10
- [ 100-26-5 ]
- neodymium(III) nitrate hexahydrate [ No CAS ]
- [ 865169-07-9 ]
- Nd(3+)*C20H11N4O2(1-)*C7H3NO4(2-) [ No CAS ]
| Yield | Reaction Conditions | Operation in experiment |
|---|---|---|
| 65% | With sodium hydroxide; In water; at 179.84℃; for 72h;pH 8 – 9;Autoclave; High pressure; | General procedure: PrCl3·7H2O (0.1mmol), HNCP (0.1mmol) and 2,5-H2pydc (0.1mmol) were mixed in 10ml deionized water. And its pH value was controlled in the range of 8-9 with 1mol·L-1 NaOH solution. The resulting precursor was sealed in 25ml Teflon-lined stainless steel reactor and heated at 453K for 3d under autogenous pressure. The reaction system was then cooled slowly to room temperature. The obtained solid is a mixture of yellow block crystals and powder. The crystals of 1 suitable for X-ray single-crystal diffraction analysis are picked out from the solid mixture in 61% yield based on PrCl3·7H2O. Syntheses of [Ln(NCP)(2,5-pydc)]n (Ln=La(2), Nd(3), Sm(4)). The complexes 2, 3 and 4 were prepared as described as for 1 above, except that Ln(NO3)3·6H2O was used instead of PrCl3·7H2O. The crystals of complexes 2-4 are picked out from the solid mixture in 67%, 65% and 72% yield based on Ln(NO3)3·6H2O, respectively. Anal. Calcd for C54H34N10O15Pr2 (1): C 48.23, H 2.55, N 10.42%. Found: C 48.01, H 2.63, N 10.10%. Anal. Calcd for C27H14N5O6La (2): C 50.41, H 2.19, N 10.89%. Found: C 50.13, H 2.33, N 10.84%. Anal. Calcd for C27H14N5O6Nd (3): C 49.99, H 2.18, N 10.80%. Found: C 50.03, H 2.25, N 10.77%. C27H14N5O6Sm (4): C 49.53, H 2.16, N 10.70%. Found: C 49.47, H 2.20, N 10.67%. |
- 11
- [ 100-26-5 ]
- samarium(III) nitrate hexahydrate [ No CAS ]
- [ 865169-07-9 ]
- Sm(3+)*C20H11N4O2(1-)*C7H3NO4(2-) [ No CAS ]
| Yield | Reaction Conditions | Operation in experiment |
|---|---|---|
| 72% | With sodium hydroxide; In water; at 179.84℃; for 72h;pH 8 – 9;Autoclave; High pressure; | General procedure: PrCl3·7H2O (0.1mmol), HNCP (0.1mmol) and 2,5-H2pydc (0.1mmol) were mixed in 10ml deionized water. And its pH value was controlled in the range of 8-9 with 1mol·L-1 NaOH solution. The resulting precursor was sealed in 25ml Teflon-lined stainless steel reactor and heated at 453K for 3d under autogenous pressure. The reaction system was then cooled slowly to room temperature. The obtained solid is a mixture of yellow block crystals and powder. The crystals of 1 suitable for X-ray single-crystal diffraction analysis are picked out from the solid mixture in 61% yield based on PrCl3·7H2O. Syntheses of [Ln(NCP)(2,5-pydc)]n (Ln=La(2), Nd(3), Sm(4)). The complexes 2, 3 and 4 were prepared as described as for 1 above, except that Ln(NO3)3·6H2O was used instead of PrCl3·7H2O. The crystals of complexes 2-4 are picked out from the solid mixture in 67%, 65% and 72% yield based on Ln(NO3)3·6H2O, respectively. Anal. Calcd for C54H34N10O15Pr2 (1): C 48.23, H 2.55, N 10.42%. Found: C 48.01, H 2.63, N 10.10%. Anal. Calcd for C27H14N5O6La (2): C 50.41, H 2.19, N 10.89%. Found: C 50.13, H 2.33, N 10.84%. Anal. Calcd for C27H14N5O6Nd (3): C 49.99, H 2.18, N 10.80%. Found: C 50.03, H 2.25, N 10.77%. C27H14N5O6Sm (4): C 49.53, H 2.16, N 10.70%. Found: C 49.47, H 2.20, N 10.67%. |
- 12
- [ 100-26-5 ]
- [ 865169-07-9 ]
- praseodymium (III) chloride heptahydrate [ No CAS ]
- [ 7732-18-5 ]
- Pr(3+)*C20H11N4O2(1-)*C7H3NO4(2-)*1.5H2O [ No CAS ]
| Yield | Reaction Conditions | Operation in experiment |
|---|---|---|
| 61% | With sodium hydroxide; at 179.84℃; for 72h;pH 8 – 9;Autoclave; High pressure; | PrCl3·7H2O (0.1mmol), HNCP (0.1mmol) and 2,5-H2pydc (0.1mmol) were mixed in 10ml deionized water. And its pH value was controlled in the range of 8-9 with 1mol·L-1 NaOH solution. The resulting precursor was sealed in 25ml Teflon-lined stainless steel reactor and heated at 453K for 3d under autogenous pressure. The reaction system was then cooled slowly to room temperature. The obtained solid is a mixture of yellow block crystals and powder. The crystals of 1 suitable for X-ray single-crystal diffraction analysis are picked out from the solid mixture in 61% yield based on PrCl3·7H2O. Syntheses of [Ln(NCP)(2,5-pydc)]n (Ln=La(2), Nd(3), Sm(4)). The complexes 2, 3 and 4 were prepared as described as for 1 above, except that Ln(NO3)3·6H2O was used instead of PrCl3·7H2O. The crystals of complexes 2-4 are picked out from the solid mixture in 67%, 65% and 72% yield based on Ln(NO3)3·6H2O, respectively. Anal. Calcd for C54H34N10O15Pr2 (1): C 48.23, H 2.55, N 10.42%. Found: C 48.01, H 2.63, N 10.10%. Anal. Calcd for C27H14N5O6La (2): C 50.41, H 2.19, N 10.89%. Found: C 50.13, H 2.33, N 10.84%. Anal. Calcd for C27H14N5O6Nd (3): C 49.99, H 2.18, N 10.80%. Found: C 50.03, H 2.25, N 10.77%. C27H14N5O6Sm (4): C 49.53, H 2.16, N 10.70%. Found: C 49.47, H 2.20, N 10.67%. |
- 13
 [ 82410-79-5 ]
[ 82410-79-5 ]- [ 100-26-5 ]
- copper(II) nitrate trihydrate [ No CAS ]
- [ 7732-18-5 ]
- [Cu2(bix)1.5(2,5-pyridine-dicarboxylate)2(H2O)]*2H2O}n [ No CAS ]

Reviews
There are no reviews yet.